
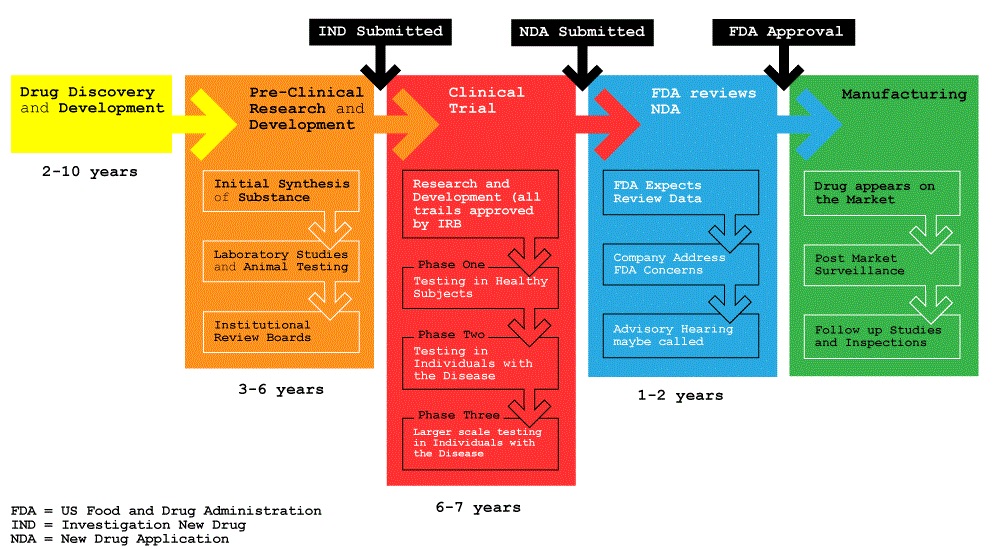
U.S. Food and Drug Administration (FDA) approval is a long process that takes an average of 12 years from development in a laboratory to becoming available in pharmacy. Drug development involves several rounds of testing, which help determine if the drug being tested is both safe and effective.
 |
| Source: SRxA |
Initially, development begins in the laboratory with molecule discovery and characterization. Further molecular biology studies are conducted, which help find more than 10,000 to 20,000 compounds. These thousands of compounds will be tried in in vitro screenings to find a few favorable candidates; the safest of these candidates will go forward in animal trials. These animal trials will help find the best compounds to be tried in a Phase I trial.
A Phase I trial is conducted in about 50 healthy subjects to test several compounds to determine their toxicity, best route of administration, and a safe dose to give. If the compound is then still considered safe, it will go onto Phase II studies.
Phase II studies have about 250 patients who will be testing the compound to determine if the compound is effective, if it can be tolerated, and safe doses for the compound. The sponsor of the compound should have doses that are expected to be higher than what the expected concentration is needed to avoid cause resistance in the virus, but also to try to show that the compound has a response based on the dose of the compound given.
Once this phase of testing is complete, Phase III testing begins in a much larger patient population of around 3,000. Phase III testing seeks to utilize the foundation of efficacy, tolerability and dosing that was determined during Phase II testing. Phase II and Phase III are often controlled studies in which the compound is also compared to other commonly used treatments and the use of the compound with other drugs. If the compound is then determined to be both safe and effective in this larger scale, setting it is then submitted for approval to the FDA; this part of the process can take two years to approve.
One additional phase exists for a drug that has entered the market; this is Phase IV or Post Market Surveillance. This phase of testing seeks to gather information from a more diverse population and to determine any side effects that can occur with long-term use of the drug. This is necessary since it is not possible to predict all of the side effects that may occur with a drug. If there are serious adverse effects that are detected during this phase, the FDA may have to take action that could limit the drug’s use.
Once a new drug has been approved by the FDA it can be prescribed by a physician. However, it is not immediately available to your local pharmacy; it usually takes a couple of weeks to find its way to the pharmacy shelf. A pharmacy may choose to wait until a patient comes in with a prescription before ordering the medication, especially if the medication is expensive.
Five out of about 5,000 compounds make it from preclinical testing to human testing and then one out of those 5 will be approved for usage in humans. It is estimated that it costs about $350 million. The FDA does not complete the clinical trials themselves; the studies are completed by the drug companies. The drug companies have to pay fees to the FDA in order to get the medication reviewed. The FDA can allow for patients to receive the medication before the approval of the medication through an expanded access program. Patients are allowed to get the medication if they meet specific criteria for need and that they do not qualify for the current clinical trial.
For more information on the FDA approval process, click here.




Comments
Comments